 TUCF Genomics |
Tufts Core Facility | Illumina |
|
[ Login ] Other TUCF Core Services
Business Hours
Monday to Friday 9:00AM to 5:00PM (Phone) 617.636.3992 (Fax) 617.636.6737 |
Frequently Asked QuestionsHow to make with credit card?How is a library created? How is a library sequenced with Illumina technology? What is the difference between “Single-End” and “Paired-End” reads? How many samples can be run on a HiSeq Flow cell? What are adapter dimers and how do I get rid of them? Does my sample have constant sequence? How does that affect the sequencing process? How to make with credit card?Due to University policy, we CANNOT receive credit card information via email or on-line. If you would like to make payments with a credit card, please phone in information or request on-line payment link. How is a library created?Below is the typical workflow for sequencing genomic DNA or cDNA. These are steps necessary to create random DNA fragments that are flanked by adapters which are required for sequencing on the Illumina HiSeq2000. 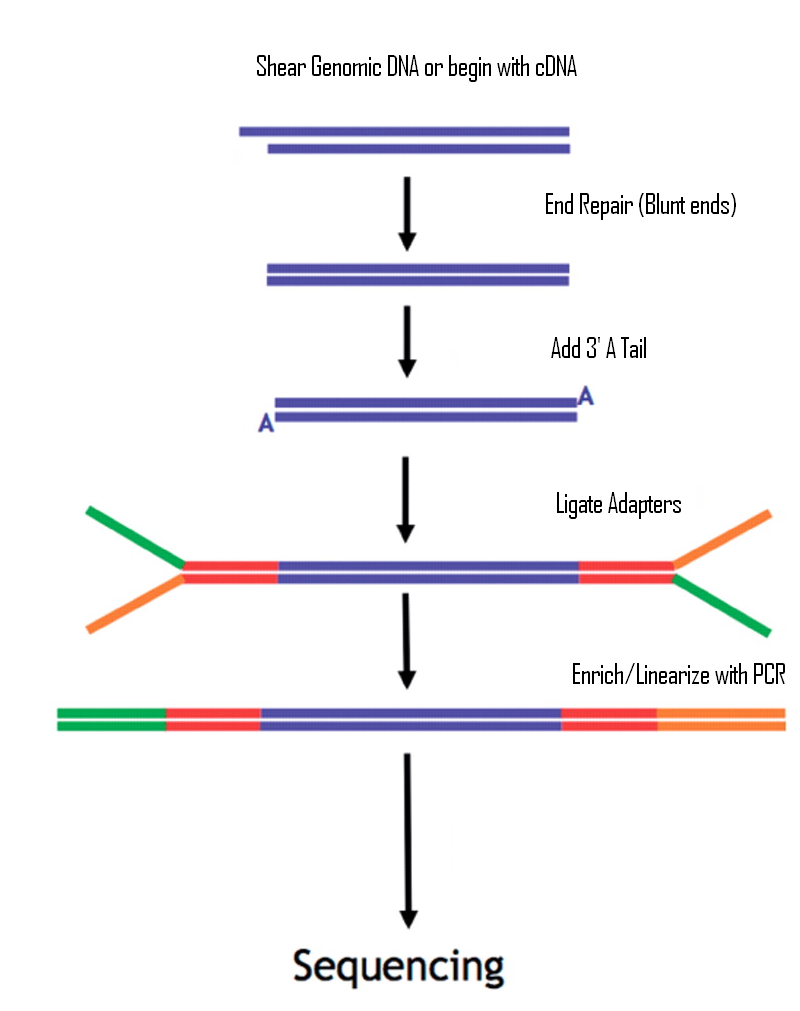 How is a library sequenced with Illumina technology?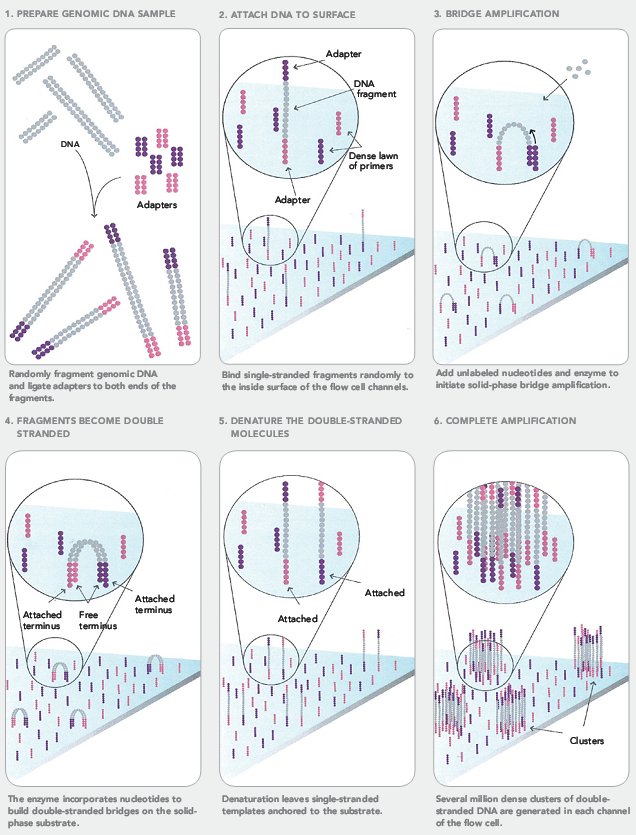 Essentially, after library preparation is completed the libraries are denatured, flown over the flow cell, and undergo a process known as “bridge amplification” in order to create clonal clusters of single stranded DNA molecules. Following this the DNA is sequenced one base at a time using Reversible Terminator chemistry (Below): 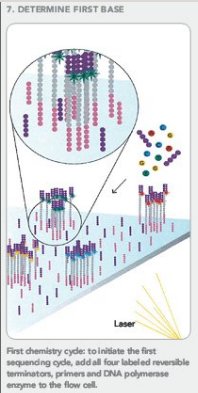 A sequencing primer binds to either the Universal or Indexing adapter (See “Understanding Illumina TruSeq Adapters”), and one base labeled with a specific fluorescent color is added. Because the clusters contain identical DNA sequences the entire cluster is read as one base. A camera records these base reads across the entire flow cell (See Below). 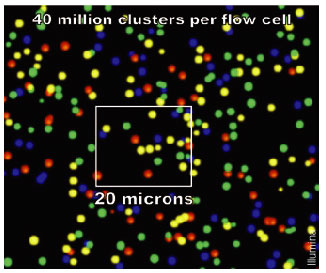 This takes place for 50 or 100 cycles to create about 150-200million 50bp or 100bp reads. What is the difference between “Single-End” and “Paired-End” reads?Single-End Read: When the sequencing process only occurs in 1 direction (utilizing Read Primer 1). Paired-End Read: If two separate read cycles occur in both directions (utilizing both Read Primer 1 and 2). This kind of read will provide data about both sides of the fragment of interest (Blue). If the fragment size is consistent you will also be able to predict that both the forward and reverse reads will be a known distance from each other. This data can be used to help the software map the reads more accurately. 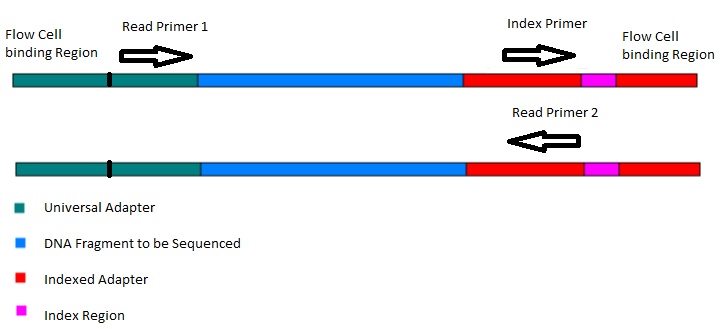 How many samples can be run on a HiSeq Flow cell?The HiSeq flow cells contain 8 separate lanes. Each one of these lanes can have as many samples mixed together as a user wishes. The “Indexed Adapter” contains a 6-8 bp region which is unique to a sample and serves as a “barcode.” The only drawback is that the amount of overall data achieved is split among the total number of samples, so the user should have a good idea of how much data is required so they don’t get too much data (waste of money) or insufficient data (will require additional sequencing). What are adapter dimers and how do I get rid of them?Adapter dimers are formed during the ligation portion of the protocol when two adapters are joined together. These are problematic because they can bind to the flow cell and undergo sequencing, but provide no data other than the sequence of the adapter present. This problem can be approached several ways:
Does my sample have constant sequence? How does that affect the sequencing process?Anytime a sample has the same bases within the “DNA fragment of interest” it is called a constant sequence. This happens when people use 5’ or 3’ barcoding, or when a protocol requires amplifying regions of DNA which have several bases that remain the same but have a variable region of interest after a certain point (such as antibody variable regions or 16s metagenomic analysis). During the first few cycles of sequencing the software identifies where each cluster is on the flow cell by analyzing signal intensity for a specific base. When constant sequence is present during this process it makes cluster identification very difficult because every cluster is read as the same base. If the intensity of the signal is too high or cluster intensities overlap it can cause the run to fail completely in a given lane. Therefore it is often prudent to load the lane at a much lower concentration so that the signal intensity can be reduced. Additionally, it is possible to load a sample with a completely random sequence to add some “complexity” (base variability) to each read and simply remove those reads in the data analysis steps. |